S příchodem internetu a svobodných médií se lidé začali o smrtelných nemocech dozvědět více a více –
Co je prionová choroba
Prionové nemoci – také se jim říkáPřenosné spongiformní encefalopatie jsou rodinou vzácných progresivních neurodegenerativních onemocnění, která postihují lidi i zvířata. Vyznačují se:
- dlouhá inkubační doba;
- charakteristické uvolnění houbovité tkáně mozku spojené se ztrátou neuronů;
- neschopnost imunitního systému reagovat na infekci spuštěním zánětlivého procesu.
Prionová onemocnění postihují lidi i zvířata, rychle postupují a vždy vedou k smrti.
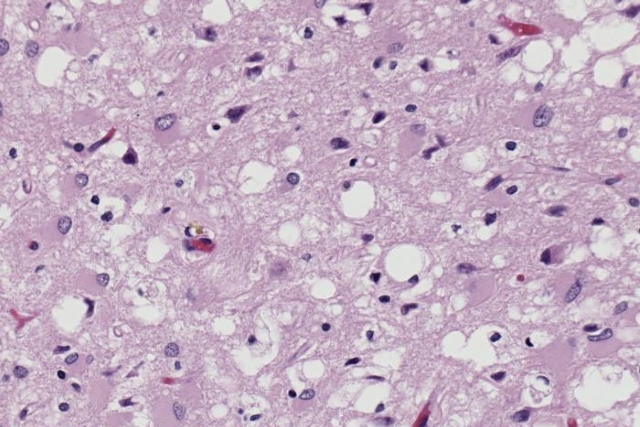 Prióny v makru
Prióny v makru
Původcem onemocnění jsou priony, typ proteinu sabnormální terciární struktura, neobsahující nukleové kyseliny. Samotný termín se týká patologických patogenních agens, která jsou schopna způsobit abnormální skládání specifických normálních buněčných proteinů, které se nazývají prionové proteiny, nacházející se nejčastěji v mozku. Funkce těchto normálních prionových proteinů stále nejsou plně pochopeny.
Creutzfeldt-Jakobova choroba— CJN, spastická pseudoskleróza, syndrom kortikostriospinální degenerace, přenosná spongiformní encefalopatie, nemoc šílených krav.
Jedná se o progresivní degenerativní onemocněnímozková kůra, bazální ganglia a mícha. Je považována za hlavní projev spongiformní encefalopatie (prionové onemocnění). Neexistuje žádný lék. CJN postihuje lidi všech národností a ras, muže i ženy, dospělé i děti.
Prionové proteiny- normální bílkoviny, které jsou přítomny vkaždý. Ale existují určité skupiny lidí, kteří mají genetickou mutaci, která je předurčuje k syntéze patogenního prionového proteinu. Prionová onemocnění se mohou přenášet i přímou infekcí, k přenosu může dojít i při operaci, užívání lidského růstového hormonu nebo konzumací kontaminovaného masa. Tento typ infekce se nazývá iatrogenní a ve srovnání s jinými formami CJN zůstává v menšině.
Procento iatrogenních případů Creutzfeldt-Jakobovy choroby v National CJD Research & Dohledová jednotka u 177 pacientů.
- Růstový hormon (somatotropin) - 53,1% (94 případů).
- Dura mater (včetně stravování) - 38,9% (69 případů).
- Gonadotropní hormon - 2,25% (4 případy).
- Neurochirurgické přístroje - 2,25% (4 případy).
- Transplantace rohovky - 1,69% (3 případy).
- Elektrody pro stereoelektroencefalografii - 1,12% (2 případy).
- Transplantace jater - 0,56% (1 případ).
Existují případy infekce, které nejsouklasifikovány pro kterýkoli ze dvou výše uvedených důvodů, v takovém případě jsou považovány za sporadické, to znamená, že vznikly spontánně a nezávisle v důsledku okolností nezávislých na genetice nebo vnějších faktorech.
Dr. Oybek Turgunhuzhaev, vedoucípokyny pro neurorehabilitaci v Interdisciplinárním rehabilitačním centru (Moskva) uvádí, že konečná diagnóza osoby s podezřením na jakoukoliv prionovou chorobu je založena na posouzení klinických příznaků a symptomů a řady podpůrných studií. Jediná metoda pro potvrzení diagnózy byla dlouhodobě elektroencefalografie. Ale protože celková citlivost této metody je omezená, užitečnost této studie byla zpochybněna.
Proč v Rusku tuto diagnózu nedávají
Prionové nemoci jsou nevyléčitelné, jsou nevyhnutelnéfatální. Navíc je problém, že pro stanovení spolehlivé diagnózy je nutné provést pitvu. Jakákoli pitva je pro patologa rizikem, protože se vyskytly případy iatrogenní infekce specialistů od zemřelých pacientů.
Na objednávku Rospotrebnadzor, že osobanemocná s prionovou chorobou musí být oznámena do dvou hodin. Zavedení takové diagnózy vede podle ruských pokynů zároveň k likvidaci veškerého vybavení, se kterým byl pacient v kontaktu. To je důvod, proč, když Medusa řekla případ jednoho z pacientů s CJD, všechny kliniky uvedly, že nemají vybavení k pozorování těchto pacientů. Ve skutečnosti je to jen způsob, jak neztratit miliony rublů, dokonce ani vyřadit stroj MRI. Pokud by se jednalo o stovky diagnóz prionové choroby (například v USA je každoročně registrováno 300 případů, možná více), pak by to byla otázka ztráty miliard rublů pro ruské nemocnice a rozpočet. To je důvod, proč diagnóza není oficiálně učiněna, lékaři o tom nechtějí mluvit, protože neexistuje žádný oficiální pokyn, že diagnózu nelze provést. V důsledku toho se ukazuje, že je nemoc, je smrt, a neexistují důvody pro příbuzné a umírající lidi.
Nikdo jim neřekne, že s největší pravděpodobností příbuzníjiž infikovaných. Nikdo nebude říkat, že nemůžete vyzkoušet syrové mleté maso nebo jíst syrové maso, zejména mozky. Také proto, že diagnóza není provedena, je možné náhodně transplantovat orgán pacienta s prionovou chorobou, čímž infikuje jinou osobu. To může také nastat přes chirurgický nástroj (tam byly takové případy, více na tomto dole).
Když jsme požádali alespoň někoho, aby nám o tom řeklprionové nemoci, téměř nikdo nebyl připraven mluvit otevřeně. Anonymně jsme si tedy povídali s neurologem v jedné z největších moskevských nemocnic. „S priony jsou dva problémy. Za prvé, pro stanovení spolehlivé diagnózy je nutné provést pitvu. I když formálně (například podle ruských směrnic) pitvu lze provést, i když za zvláštních podmínek. Jakákoli pitva je pro patology přirozeně dalším rizikem, protože byly popsány případy, kdy se patologové nakazili od zemřelých pacientů. nikdo na ně nechce přenést riziko.
Za druhé, protože prionové infekce jsou těžképlynulé, nevyléčitelné nemoci (i když s poměrně komplikovaným způsobem přenosu) jsou v naší zemi zatraceně komplikované zákony pro registraci a řízení takových pacientů; případy musí být hlášeny v případě odhalení do dvou hodin, po diagnóze je nutné provést zničení části drahého vybavení, které, jak se může ukázat, ani leželo vedle pacienta, vydávat dokumenty a tak dále.
Docela smutné, ale přesto pravdivé, faktem je, že správná diagnóza nezáleží na pacientovi. Stále neexistuje žádný lék. “
Creutzfeldt-Jakobova choroba
Creutzfeldt-Jakobova nemoc (CJD) je jednou z nichtypů prionových onemocnění. Jde o rychle progresivní, fatální neurodegenerativní onemocnění, o kterém se předpokládá, že je způsobeno abnormální izoformou prionového proteinu. CJD se vyskytuje po celém světě a statistiky odhadují, že celosvětově je postižen 1 z milionu lidí.
Prionová onemocnění nejdou stejným způsobem.Ve stejném scénáři se mohou lidé trpící stejnou prionovou lézí lišit od epidemiologie a patogeneze. Creutzfeldt-Jakobova choroba je rozdělena do několika typů.
Sporadická forma - bolest z mutace, kašel, bolest hlavy a paměť
Sporadická Creutzfeldtova-Jakobova choroba (SCAN) -nejběžnější typ přenosné spongiformní encefalopatie u lidí, který představuje přibližně 85% případů registrovaných onemocnění prionové povahy. SBCYA má velmi rychlý průběh onemocnění - průměrná délka života po nástupu symptomů je pouze šest měsíců. Více než 90% pacientů zemře do jednoho roku od nástupu příznaků. K nejvyššímu výskytu dochází u starších osob ve věku 60–70 let, v jiných věkových skupinách se to děje mnohem méně často. Jedním z předpokladů vzniku SCDM je názor, že se jedná o spontánní neurodegenerativní onemocnění vyplývající ze somatické mutace PRNP genu nebo náhodná strukturální změna PrP proteinu, která způsobuje tvorbu PrPSc2. Epidemiologické studie neodhalily souvislost mezi sporadickou formou CJD a faktory prostředí.
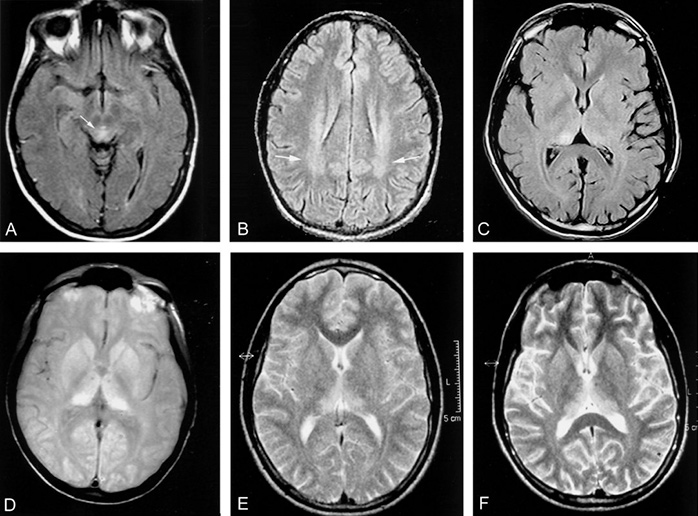 Snímek mrt pacient CJD
Snímek mrt pacient CJD
První příznaky SCAD jsou obvykle nespecifické: bolest hlavy, malátnost, kašel, závratě a změny v chování, náladě nebo přerušení paměti. Pro potvrzení diagnózy musí uplynout čas, aby se z jiných důvodů objevila prionová povaha. Klasické klinické příznaky CJD jsou:
- rychlý pokles kognitivních schopností;
- ataxie (porušení koordinace pohybu různých svalů);
- myoklonus (rychlé náhlé kontrakce jednotlivých svalů), končící akinetickým mutismem (inhibice všech motorických funkcí, kromě fixačních pohybů očních bulbů).
Konečná diagnóza závisí na vyhodnocení klinických projevů a výsledků laboratorních testů.
Akinetický mutismus- stav, kdy se pacienti zastavípohybují se a sledují cíl očima, s výjimkou reakce očí na podněty nebo delší fixace pohledu, jejich svaly se periodicky rychle stahují samy nebo pod vlivem vnějších faktorů. Pacienti trpí inkontinencí a nevydávají žádné zvuky nebo pouze artikulované zvuky. Pokud polykání přetrvává, pacienti mohou žít v tomto stavu několik týdnů, dokonce i let s jinými příznivými faktory, přičemž výživu dostávají nitrožilně nebo sondou. U sporadické formy CJN dosáhnou pacienti tohoto stavu během prvních týdnů onemocnění. V nejrychlejších scénářích dosáhne 10 % pacientů tohoto stavu do jednoho roku.
Dlouhou dobu nejinformativnějším způsobemdiagnóza byla provedena pomocí difuzně-tenzorové MRI. Tato metoda je nejpřístupnější, relativně neinvazivní a účinná pro časné změny mozkové kůry. SBKYa může být detekován pomocí markerů v nosních sliznicích, mozkomíšním moku, moči nebo krvi, ale tyto testy často dávají falešně pozitivní výsledky - protein 14.3.3 není specifický bez doprovodného klinického obrazu. Proteinogram je nová optimální metoda pro diagnostiku SCD, protože je nejcitlivější ze všech výše uvedených.
Proteiny 14-3-3- nalezená rodina regulačních molekulu všech eukaryot. Vážou se na řadu dalších proteinů, regulují jejich funkce a tím ovlivňují mnoho procesů, včetně regulace buněčného cyklu, metabolické kontroly, apoptózy a kontroly genové transkripce. Byly objeveny před více než 40 lety při systematické klasifikaci bílkovin v nervové tkáni, kde jejich obsah přesahuje 1 % všech bílkovin. Dosud bylo popsáno více než 300 různých cílových proteinů schopných interakce s 14-3-3.
Proteinogram- studie, která studuje kvantitativní vztah proteinových odrůd v krvi. Pojem celkový protein zahrnuje všechny možné proteiny, navzdory jejich rozdílům ve struktuře a funkci.
Dědičná forma Creutzfeldt-Jakobovy chorobya fatální rodinná insomnie spojená s genetickými mutacemi představuje pouze 10% všech případů prionových onemocnění. Mnoho studií ukazuje, že obecná cesta v patogenezi nemoci může být běžná jak pro sporadické, tak dědičné formy prionové choroby, s výjimkou toho, že v prvním případě dochází k transformaci proteinu bez účasti jakýchkoli faktorů a není předurčena přítomností mutace. v genech.
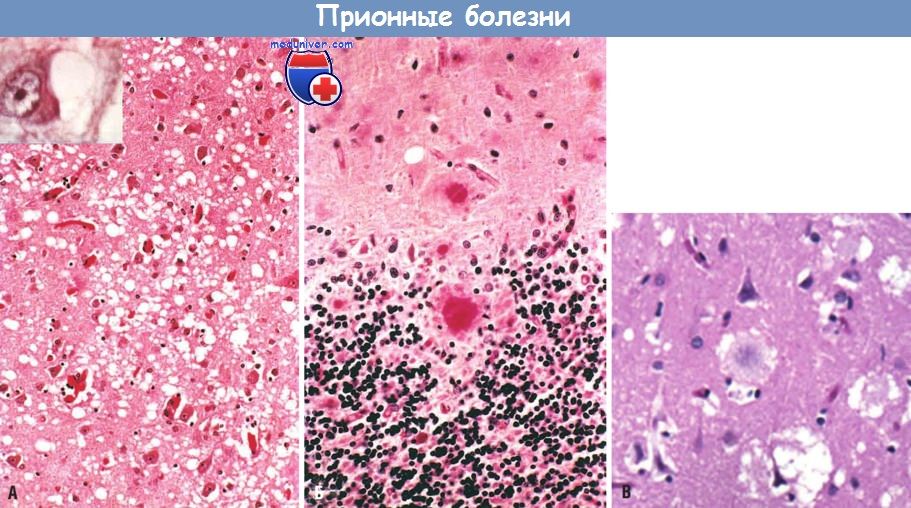 (A) Houbovitá degenerace v kortexu velkýchhemisféry. Inset: neurony obsahující vakuoly. (B) Cerebellumová kůra s kuru plaky (PAS reakce), která se skládá z PrPsc agregátů. (B) Plaky v kortikální látce, obklopené degenerační zónou ve tvaru spongy, s novou variantou Creutzfeldt-Jakobovy choroby.
(A) Houbovitá degenerace v kortexu velkýchhemisféry. Inset: neurony obsahující vakuoly. (B) Cerebellumová kůra s kuru plaky (PAS reakce), která se skládá z PrPsc agregátů. (B) Plaky v kortikální látce, obklopené degenerační zónou ve tvaru spongy, s novou variantou Creutzfeldt-Jakobovy choroby.
Studium dědičných forem CJD a fatálníchrodinná insomnie poskytla vědcům možnost studovat průběh fenotypové heterogenity (různé „kmeny“) prionové choroby. Ačkoli mnoho jiných neurodegenerativních onemocnění, jako je Alzheimerova choroba, amyotrofická laterální skleróza a Huntingtonova chorea, jsou ve fenotypu velmi podobné, prionová choroba zahrnuje mnoho klinicky odlišných symptomů.
Fenotyp- soubor vlastností vlastní určité fázi vývoje onemocnění. Fenotyp se tvoří na základě genotypu.
Polymorfismus- schopnost některých organismů existovat ve stavech s různou vnitřní strukturou nebo v různých vnějších formách.
S dědičnou prionovou chorobou, fenotypemonemocnění bude určeno kombinovaným účinkem patogenních mutací, polymorfismu kodonu 129 a typu PrP Sc. Polymorfismus Codon 129 hraje dvojí roli v predikci výsledku onemocnění. Ústředním bodem pro pochopení patogeneze prionového onemocnění je detailní a přesná znalost procesů a podmínek in vivo pro tvorbu PrPSc, což nevyhnutelně vede k vývoji a expresi onemocnění. Tyto znalosti vám umožní vyvinout racionální a účinnou strategii pro terapeutickou intervenci.
Nová varianta CJD - nakažení nakažením kusu masa
Nová varianta Creutzfeldt-Jakobovy choroby(nvCJD) byl poprvé identifikován v roce 1996. Následné studie podpořily hypotézu, že tato forma je spojena s bovinní spongiformní encefalopatií. S největší pravděpodobností pacienti jedli maso obsahující patologické priony kravského mozku.
Na rozdíl od sporadické formy toto onemocnění nenímá jasný věk infekce. Pacienti s nCJN často vykazují také psychiatrické symptomy, a proto je někdy mylně diagnostikována spíše jako duševní porucha než jako neurologická porucha. Pravá příčina psychiatrických symptomů spočívá v kognitivních poruchách, neustálých bolestech končetin, poruchách přiměřenosti čití (parestézie nebo dysestézie), poruchách řeči nebo zraku.
Vady se vyvinou během 6–8 měsícůovládání svalového systému, ale v některých případech může vývoj onemocnění trvat déle než 18 měsíců. Proto je při prvních příznacích onemocnění poměrně obtížné stanovit tuto diagnózu. Pokud pacient zažívá nekontrolované pohyby, zvyšuje se pravděpodobnost správné diagnózy nCJN. Na rozdíl od sporadické formy, která se vyznačuje náhlými svalovými křečemi při napětí (myoklonus), v případě nové formy dochází jak k dystonii (syndrom, při kterém dochází k neustálému křečovitému stahování svalů), tak k choree (syndrom charakterizovaný nepravidelnými, trhavými, nepravidelné pohyby) jsou možné.
Smrtelné stadium nové formy je podobné smrtelnémuStádium sporadické Creutzfeldt-Jakobovy choroby se vyskytuje s progresivní ztrátou svalové kontroly, což často vede ke stavu akinetického mutismu.
Demence a CJD: někdo je nemocný a o tom ani neví
Na rozdíl od běžnějších slabomyslnýchstavy, jejichž vývoj obvykle trvá roky, rychle postupující demence se mohou vyvinout během měsíců, týdnů nebo dokonce dnů a vést ke smrti. Sporadická forma CJN tvoří 46,9 % všech registrovaných případů rychle progredující demence a genetická forma prionových onemocnění tvoří 13,6 %. Frontotemporální demence (FTD), kortikobazální degenerace (CBD), Alzheimerova choroba (AD), demence s Lewyho tělísky (DLB) a progresivní paralýza tvoří 39 % všech případů.
Zpravidla je prezentována sporadická forma CJNkombinace demence a neurodegenerativních nebo psychiatrických symptomů. Pyramidální, cerebelární a fokální kortikální dysfunkce jsou u takových pacientů běžné. U třetiny pacientů předcházejí demenci stížnosti na únavu, bolesti hlavy, poruchy spánku, malátnost, hubnutí, bolesti, deprese nebo změny chování.
Neurologické příznaky, včetně ataxie,dysestézie, demence nebo svalové poruchy (chorea, myoklonus nebo dystonie) se objevují později. Většina případů rychle progredující demence bez dalších přidružených příznaků se vyskytuje u starších lidí v důsledku metabolických poruch nebo akutních infekcí (pneumonie nebo infekce močových cest). Lékaři proto před absolvováním laboratorních testů indikují především rychle postupující demenci bez jasné diagnózy. Konečný verdikt bude záviset na výsledcích testů a vyšetření, nemoci se budou lišit v závislosti na klinickém obrazu. EEG může pomoci vyloučit aktivitu záchvatů v mozku a pomoci diagnostikovat další stavy, jako je CJD.
Rychle se rozvíjející demencejeden z nejtěžších neurologických problémů. Diferenciální diagnóza je široce používána pro potvrzení definitivních diagnóz, které se mohou týkat neurodegenerativních, autoimunitních, infekčních a neoplastických onemocnění. I s takovým pečlivým přístupem k vyšetření pacientů je malé procento případů diagnostikováno po smrti.
Fatální familiární insomnie
Fatální rodinná insomnie je vzácnáprionové onemocnění, které vás doslova připravuje o spánek a vede k poklesu všech neuromotorických a mentálních funkcí. Existují dvě formy tohoto onemocnění: genetické a sporadické.
Genetická forma je spojena s mutací vedoucí kke konverzi PrP proteinu na prionový protein. Sporadicky se objevuje spontánně, bez jakýchkoliv předpokladů. Toto onemocnění se od ostatních prionových onemocnění liší v oblasti poškození – k přeměně prionových proteinů na patologické dochází především v jedné části mozku – thalamu, který je mimo jiné zodpovědný za spánek.
Průměrný věk nástupu příznakůgenetická forma smrtelné rodinné nespavosti - 40 let. Mezi časné příznaky patří menší potíže s usínáním a setrváním ve spánku, svalové záškuby, křeče a necitlivost. Během spánku mohou lidé spát neklidně: pohybovat nohama a rukama, házet a otáčet. Nakonec pacienti přestanou spát. Kvůli tomu se snižuje mentální aktivita a ztrácí se svalová koordinace (ataxie). Pacienti pociťují zvýšený krevní tlak, zvýšenou srdeční frekvenci a nadměrné pocení. Na rozdíl od jiných prionových onemocnění nedochází u fatální familiární nespavosti ke spongiformní degeneraci mozkové substance. Hlavními příznaky poškození jsou neuronální degenerace a reaktivní glióza (tvorba „jizev“ na a v mozkové tkáni) v jádrech thalamu, stejně jako neuronální degenerace v komplexu medulla oblongata nuclei.
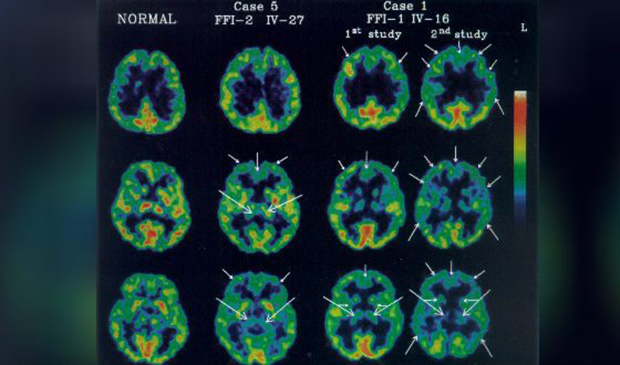 Mozek nemocný s fatální rodinnou nespavostí
Mozek nemocný s fatální rodinnou nespavostí
Diagnóza fatální familiární insomnie vgenetická forma je potvrzena genetickým vyšetřením. Ve sporadických případech mohou být poruchy spánku a abnormality v thalamu detekovány polysomnografií a pozitronovou emisní tomografií (PET). Průměrná délka života od propuknutí prvních příznaků onemocnění je 3 roky, léčba neexistuje.
Pomalu postupující demence: symptomy během několika let
Gerstmann-Straussler-Scheinkerův syndrom jevzácná genetická forma přenosné spongiformní encefalopatie, kterou poprvé popsali rakouští neurologové v roce 1936. Nejčastěji se syndrom objevuje mezi 40. a 50. rokem života a je způsoben mutacemi genů prionového proteinu PRNP na 20. chromozomu.
Klinický obraz syndromu je podobnýsporadická forma CJD, ale liší se délkou trvání a pomalu progresivní demencí spolu s dalšími symptomy. Gerstmann-Straussler-Sheinkerův syndrom může trvat několik měsíců nebo několik let, průměrná délka života je 5 let. Je možné diagnostikovat nemoc i v raných fázích pomocí magnetické rezonance. MRI bude vykazovat houbovité změny v kortexu a proliferaci gliálních buněk.
Gliální buňky nebo neuroglia- soubor pomocných buněk nervové tkáně. Tvoří asi 40 % objemu centrálního nervového systému. Počet gliových buněk v mozku se přibližně rovná počtu neuronů.
Gliové buňky mají společné funkce a zčástipůvodu (výjimka - mikroglie). Představují specifický mikroprostředí neuronů, zajišťují podmínky pro generování a přenos nervových impulzů, jakož i provádění části metabolických procesů samotného neuronu.
Detekce prionové choroby je dostačujícíVzhledem k tomu, že je-li diagnóza potvrzena, pacient již není schopen pomoci a nemocnice ztrácejí miliony rublů. A nejhorší je, že jsou ohroženi všichni lidé na Zemi. Prions nikoho nezachrání. Nepokoušejte se proto náplň po solení. Nejezte střeva a mozky krav a prasat a včas navštivte lékaře. Nakonec MRI nebude ležet.